Адам геномында шамамен 30 мың ген бар. Бірақ олардың біреуі зақымдалса да, қорқынышты салдарға әкелуі мүмкін. Ал 23 жұптың ішіндегі бір ғана хромосом туралы айтуға не дерсіз!
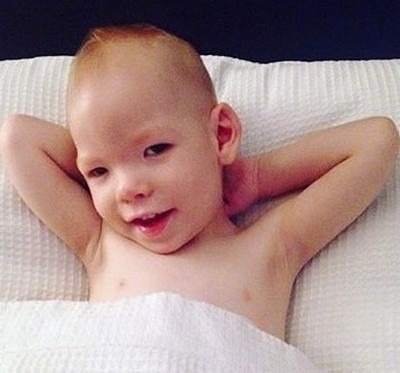
Бұл синдром 5-хромосоманың мутациясымен (5р-). Алғаш рет француз педиатры Жером Лежён 1963 жылы сипаттап, оның атымен аталған. Айтпақшы, Дауна синдромын түсіндіруге дәл осы Лежён 21 жұптағы артық хромосоманы тапқан.
Мұндай ауытқуы бар балалар туылғанда мысықтың мияулауына ұқсайтын ерекше айқай шығарады. Бірақ «мысықтық жылау», өкінішке орай, аурудың ең зиянсыз көрінісі болып табылады. Бұл кеңірдектің өзгеруімен байланысты (тарылуы, шеміршектердің жұмсақтығы, эпиглоттың кішіреюі, шырышты қабықтың ерекше бүктелуі). Негізінен Лежон синдромы — бұл терең ақыл-есі және дене кемістігі.
Мысықтың жылау синдромы бар балалардың сыртқы келбеті сипаттамалы: ай тәрізді бет, микроцефалия, антимонголоид көз қиықтары, қылилық, биік таңдай, тегіс мұрын. Құлақ қалқандары деформацияланған және төмен орналасқан. Сонымен қатар, туа біткен жүрек ақаулары және кейбір басқа ішкі органдардың ақаулары, жүре алмайтындық, бұлшық ет әлсіздігі кездеседі.
Көптеген науқастар өмірінің алғашқы жылдарында қайтыс болады.
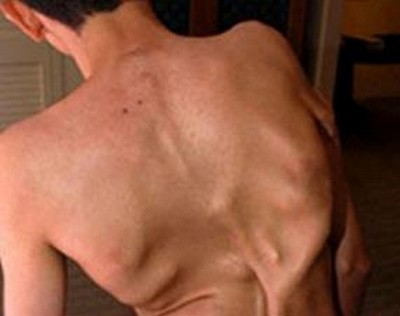 Қатайған ауру
Қатайған ауруҒылыми атауы - прогрессивті оссификацияланатын фибродисплазия (ФОП). Бұл туа біткен патология ACVR1/ALK2 ген мутациясының салдарынан пайда болады және жер шарындағы екі миллион тұрғынның біреуінде кездеседі.
ФОП ауруымен зардап шегетін адамдарда бұлшықеттерде, байламдарда, сіңірлерде және басқа байланыстырғыш тіндерде болатын жарақаттар немесе қабыну процестері осы тіндердің сүйекке айналуына және кальцификациялануына әкеледі. Фибродисплазияны тағы «екінші қаңқа ауруы» деп те атайды: ақаулы ген сүйектердің қажетсіз жерлерде тоқтаусыз өсуін (оссификаттарды) тудырады.
Генетикалық ауру 10 жасқа дейінгі жаста басталады. Баланың арқасында жиі төбешіктер пайда болады. Дәрігерлер әдетте онкологияға күмәнданады, диагноз қойылғанша көптеген талдаулар мен зерттеулер жасалады. Сонымен қатар кез келген ине салу немесе көгеру оссификатаның өсуін тудыруы мүмкін.
Ауру әртүрлі науқастарда әртүрлі жылдамдықпен өтеді. Толық қозғалмау және ерте өлім қаупі бар.
Соңғы кездері ФОП науқастары үшін үміт пайда болды: 2006 жылы ғалымдар қай генетикалық ауытқу «екінші қаңқа»ның пайда болуына әкелетінін анықтады, ал қазір патологиялық тіннің өсуін тоқтататын препарат сынақтан өтуде.
 Прогерия
ПрогерияСирек кездесетін, бірақ кеңінен танымал генетикалық ақау - организмнің ерте қартаюы. Негізгі формалары - бала жасындағы прогерия – Гетчинсон-Гилфорд синдромы және ересектердің прогериясы – Вернер синдромы.
Ересектердің прогериясы (ақаулы ген — WRN) тері мен бұлшықеттердің қарттық өзгерістерімен, катаракта дамуымен, диабеттің, атеросклероздың және басқа да «қарттық» аурулардың дамуымен көрінеді. Жыныстық жетілу кезеңінде байқалады, көбінесе ер адамдарда жиі кездеседі. 20 жастан кейін олардың шашы ағарып, түсе бастайды, қолдары мен аяқтары жіңішкеріп қалады.
Балалар прогериясы пропорционалды кішкентайлықпен, теріасты май қабатының болмауымен және сүйектердің сынғыштығымен сипатталады. Балалар прогериясының себебі — LMNA генінің мутациялары. Балалар прогериясы туа біткен болуы мүмкін болса да, көпшілігінде белгілер әдетте өмірінің екінші-үшінші жылында пайда болады.
Баланыњ ґсуі тездеп баяулайды, терісі, әсіресе беті мен ќолдарында. Ауру адам ќарќынды сыртќы кӛрініс алыды: ᴜлкен басы, мањдай бӛгірлері кішкене ютқарлы џәнімен (бір сөзбен айтқанда «ќўс тᴜріндегі») кіші бетімен мөрендірілі қараләрн џәнімен мойны, тӛменгі жағы толыќ жетілмеген. Балшыќтары атрофиятанады, тирмдерде, шаштарда, тырнаќтарда, балар мен болшыќтарда дегенеративтік процесстер жᴜреді.
Емі жоқ, науқастар әдетте атеросклероздан немесе онкологиялық аурулардан қайтыс болады.
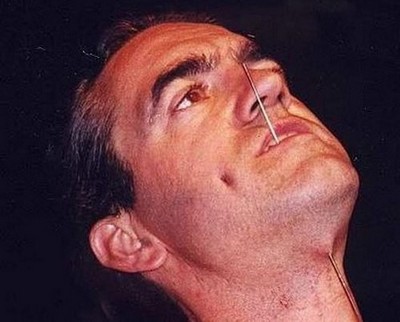 Ауырсынуды сезбеушілік
Ауырсынуды сезбеушілікБұл ауру «сенсорлық полинейропатия» деп аталады. NGFB генінің мутациясы ауырсыну рецепторларының сезімталдығының жойылуына әкеледі.
Науқастар ауырсынуды сезінбейді, бірақ дәмді, жанасуды, қысымды және дірілді сезіне алады. Яғни, олар жұмсақ сипауды сезеді, ал күшті соққыны сезбейді!
Бұған қоса, температураны қабылдау өзгереді, сондықтан адам суық пен ыстықты дұрыс қабылдай алмайды, бұл өрттер мен үсіктерге әкеледі.
Мұндай пациенттерде әлі бала кезінен бастап Шарко ауруы тәрізді буындардың бұзылуы анықталады. Бұл жүрістің бұзылуына, адамның күрделі қозғалыстарды орындай алмауына және т.б. әкеледі.
Басқа түрдегі полиневропатиялардан айырмашылығы, бұл мутациямен ауыратын балалар интеллектуалды дамуда артта қалмайды. Сонымен қатар, оларда бұлшықет атрофиясы дамымайды.
Емдеу тиімсіз, әдетте гормоналды препараттар қолданылады.
Ауру — бұл ағзаның қорғаныс реакциясы, қандай да бір проблемалардың сигналы, сондықтан ауырсыну сезімдерін толық жоғалту — бұл пайда емес. Науқастар үнемі өздеріне зиян келтіру қаупі бар — тілін тістеп алу, аяғын қайырып алу немесе сындырып алу, жарақат алу және т. б.
Шығыста кейбір адамдар, сезгіштіксіз туған, денелерін тесіп немесе басқа жарақаттар келтіріп, көше шоуларын жасап, күнкөріс табады.
Қайғы-қасірет сезімін сезбеу ерекшелігі кинематографияда белсенді қолданылады: 2012 жылы испан-француз-португал фильм «Сезімтал емес» шықты, онда осындай балалар тобы туралы айтылды.
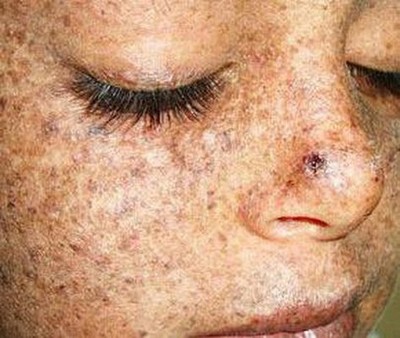 Пигменттік ксеродерма
Пигменттік ксеродермаБасқа атауы - ретикулярлық прогрессиялы меланоз, прогрессиялы ретикулярлық меланоз Пика. Бұл күн сәулелеріне - ультракүлгінге және радиацияға гиперсезімталдық. Миллион адамның шамамен төртеуінде кездеседі. Ультракүлгін сәулелену әсерінен пайда болатын ДНҚ зақымдануларын түзетуде жауапты белоктардың мутациясы себепті пайда болады.
Бірінші белгілері әдетте үш жасқа дейінгі жаста пайда болады. Тіпті күн астында бірнеше минут тұрғаннан кейін балада қатты күйіктер пайда болады. Бара-бара ашық тері аймақтары дақтармен, сепкілдермен жабылып, тері қабығылай бастайды.
Бірнеше жылдан кейін тері әртүрлі болады — кейбір жерлерде атрофия белгілері көрінеді, басқа жерлерде қараяды, тамырлы жұлдызшалар көрінеді. Сүйелдер, қабыршақтар, жарықтар, жаралар пайда болады. Мұрын, құлақ, ауыз деформацияланатын болады, көздер қабынудан және жас ағудан зардап шегеді.
Аурулар үнемі жарыққа сезімталдықты төмендететін препараттарды қабылдап, майлар мен кремдерді қолдануға, сүйел тәрізді өскіндерді жоюға мәжбүр.
Жасөспірім жасқа келгенде, терідегі түзілістер көбінесе қатерлі ісікке айналады. Науқастардың үштен екісі 15 жасқа дейін қайтыс болады.
Бұл генетикалық ауруды кинематографшылар өте «жақсы көреді». Ксеродермамен ауыратын кейіпкерлер кем дегенде 10 фильмде кездеседі.
